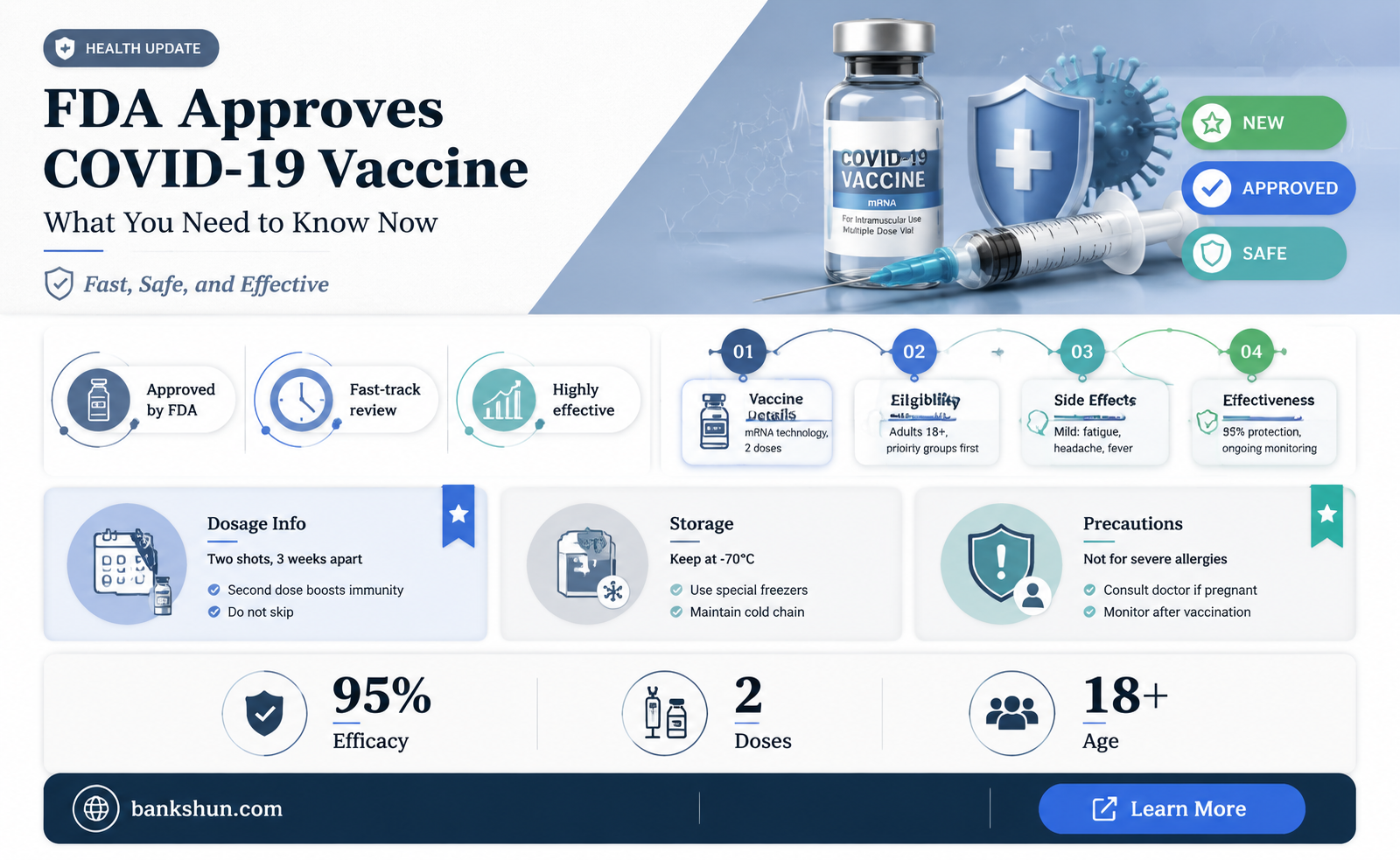
The question of whether the FDA has approved a coronavirus vaccine is a critical one, as it directly impacts public health and safety. As of the latest updates, the FDA has granted full approval to several COVID-19 vaccines, including Pfizer-BioNTech’s Comirnaty for individuals aged 16 and older, following rigorous evaluation of safety, efficacy, and manufacturing data. This approval goes beyond the initial Emergency Use Authorization (EUA) and signifies a comprehensive review process, providing additional confidence in the vaccines’ long-term benefits and risks. Other vaccines, such as Moderna’s Spikevax, have also received full approval for specific age groups, while others remain under EUA for certain populations. The FDA’s decisions are based on extensive clinical trial data and ongoing monitoring, ensuring that approved vaccines meet stringent standards for protecting against COVID-19.
| Characteristics | Values |
|---|---|
| FDA Approval Status | Fully approved (Comirnaty by Pfizer-BioNTech for ages 16+). |
| Emergency Use Authorization (EUA) | Granted for Pfizer-BioNTech, Moderna, and Johnson & Johnson vaccines. |
| Approved Age Groups | Pfizer-BioNTech: 5+ years; Moderna: 6+ years; J&J: 18+ years. |
| Vaccine Types | mRNA (Pfizer, Moderna), Viral Vector (J&J). |
| Dosage Regimen | Pfizer: 2 doses (3 weeks apart) + boosters; Moderna: 2 doses (4 weeks apart) + boosters; J&J: Single dose + booster. |
| Efficacy Rate | Pfizer: ~95%; Moderna: ~94%; J&J: ~66-72% (varies by region). |
| Common Side Effects | Pain at injection site, fatigue, headache, muscle pain, fever. |
| Approval Date (Comirnaty) | August 23, 2021. |
| Booster Recommendations | Recommended for all eligible individuals after initial series. |
| Storage Requirements | Pfizer: Ultra-cold (-90°C to -60°C); Moderna: -25°C to -15°C; J&J: 2°C to 8°C. |
| Manufacturers | Pfizer-BioNTech, Moderna, Janssen (Johnson & Johnson). |
| Latest Updates (as of 2023) | Updated boosters targeting Omicron subvariants (BA.4/BA.5) available. |
Explore related products

$2.99 $24.95





What You'll Learn
![]()
FDA's Emergency Use Authorization (EUA) process for COVID-19 vaccines
The FDA's Emergency Use Authorization (EUA) process became a critical tool in the fight against COVID-19, allowing rapid access to vaccines while maintaining safety standards. Unlike full approval, which requires extensive long-term data, EUA is granted based on preliminary evidence of safety and efficacy during public health emergencies. For COVID-19 vaccines, this meant evaluating data from large clinical trials involving tens of thousands of participants, ensuring the benefits outweighed the risks in the midst of a global pandemic.
To qualify for EUA, vaccine manufacturers had to meet specific criteria. For instance, the Pfizer-BioNTech vaccine’s EUA application included data showing 95% efficacy in preventing symptomatic COVID-19 in individuals aged 16 and older, with a two-dose regimen administered 21 days apart. Similarly, Moderna’s vaccine demonstrated 94.1% efficacy with a two-dose schedule given 28 days apart. These vaccines were authorized for emergency use in December 2020, providing a lifeline as cases surged worldwide. The FDA’s decision was based on a risk-benefit analysis, considering the urgent need for protection against a deadly virus.
One key aspect of the EUA process is its flexibility. The FDA can update authorizations as new data emerge, such as expanding eligibility to younger age groups. For example, Pfizer’s vaccine initially authorized for ages 16 and up, later received EUA for adolescents aged 12–15 in May 2021, and eventually for children as young as 5 in October 2021. This adaptability ensured that vaccines could reach vulnerable populations swiftly while maintaining rigorous safety standards.
However, EUA is not without limitations. It is a temporary measure, and manufacturers are required to continue gathering data for full approval. For instance, Pfizer’s full FDA approval in August 2021 for individuals aged 16 and older followed months of additional data collection, including real-world evidence of vaccine effectiveness and safety. This transition from EUA to full approval reinforced public confidence in the vaccine’s long-term benefits.
Practical tips for understanding EUA include recognizing that it is a legal mechanism, not a shortcut. Vaccines authorized under EUA undergo the same scientific scrutiny as fully approved vaccines, but with expedited timelines. Individuals should stay informed through trusted sources like the FDA’s website, which provides detailed fact sheets and updates on vaccine authorizations. Knowing the difference between EUA and full approval can help make informed decisions about vaccination, especially for those with concerns about safety or efficacy.
Exploring the Key Functions and Roles of Private Banks
You may want to see also
Explore related products






![]()
Pfizer-BioNTech vaccine approval timeline and criteria
The Pfizer-BioNTech COVID-19 vaccine, known as Comirnaty, became the first COVID-19 vaccine to receive full approval from the U.S. Food and Drug Administration (FDA) on August 23, 2021. This milestone followed an Emergency Use Authorization (EUA) granted in December 2020, which allowed its distribution during the pandemic’s peak. The approval process was rigorous, requiring comprehensive data on safety, efficacy, and manufacturing quality. For individuals aged 16 and older, this approval provided assurance that the vaccine met the FDA’s gold standard for long-term use, distinct from the temporary EUA status.
To achieve full approval, Pfizer-BioNTech submitted extensive clinical trial data involving over 44,000 participants. The trials demonstrated 91.3% efficacy in preventing COVID-19, with consistent results across age groups, ethnicities, and comorbidities. Safety data from millions of administered doses further supported the vaccine’s profile, with rare side effects such as myocarditis occurring primarily in young males after the second dose. The FDA’s criteria included at least six months of follow-up data to assess long-term outcomes, a requirement that distinguished full approval from the expedited EUA process.
Practical considerations for recipients include a two-dose regimen, administered three weeks apart, with a 30-microgram dose per shot. For those aged 5 to 15, a lower 10-microgram dose is used under EUA, though full approval for this age group is pending. Booster doses, typically given five months after the second shot, are recommended to maintain immunity, especially against emerging variants. Storage requirements are stringent, necessitating ultra-cold temperatures (-90°C to -60°C) for distribution, though thawed vials can be stored in standard refrigerators for up to five days.
Comparatively, the Pfizer-BioNTech vaccine’s approval timeline was expedited without compromising safety standards. While typical FDA approvals take years, the urgency of the pandemic allowed for a prioritized review, leveraging real-world data and continuous monitoring. This approach balanced speed with rigor, ensuring public trust in the vaccine’s integrity. The approval also set a precedent for other COVID-19 vaccines, such as Moderna’s Spikevax, which followed a similar path to full authorization.
For healthcare providers and the public, understanding this timeline and criteria is crucial. It underscores the vaccine’s reliability and informs decision-making, particularly for hesitant individuals. The FDA’s transparency in publishing trial data and inspection reports further reinforces confidence. As new variants emerge, ongoing studies and potential reformulations will continue to shape the vaccine’s role in pandemic management, making its approval a dynamic, evolving process rather than a static achievement.
Essential Bank Documents: Understanding the Various Types and Their Uses
You may want to see also
Explore related products






![]()
Moderna vaccine approval and safety data review
The Moderna COVID-19 vaccine, known as mRNA-1273, received emergency use authorization (EUA) from the FDA in December 2020, marking a pivotal moment in the global fight against the pandemic. This approval was based on a rigorous review of safety and efficacy data from clinical trials involving over 30,000 participants. The vaccine demonstrated 94.1% efficacy in preventing symptomatic COVID-19, with no significant safety concerns identified during the trial period. This authorization allowed for the immediate distribution and administration of the vaccine to individuals aged 18 and older, providing a critical tool to curb the spread of the virus.
Analyzing the safety data, the FDA’s review highlighted that the most common side effects were mild to moderate, including pain at the injection site, fatigue, headache, and muscle pain. These symptoms typically resolved within a few days and were more frequent after the second dose. Rarely, severe allergic reactions (anaphylaxis) were reported, prompting the FDA to recommend monitoring recipients for 15 minutes post-vaccination. Importantly, the vaccine’s safety profile was consistent across diverse demographic groups, including older adults, who are often at higher risk for severe COVID-19 outcomes.
A key aspect of the Moderna vaccine’s approval was its unique mRNA technology, which instructs cells to produce a harmless piece of the virus’s spike protein, triggering an immune response. This innovation allowed for rapid development and scalability, but also required careful scrutiny. The FDA’s review included an assessment of the vaccine’s stability and storage requirements, noting that it must be stored at -20°C (-4°F), which posed logistical challenges compared to other vaccines. Despite this, the Moderna vaccine became a cornerstone of vaccination campaigns worldwide.
For practical application, the Moderna vaccine is administered in two doses, 28 days apart, with each dose containing 100 micrograms of mRNA. In August 2021, the FDA amended the EUA to include a third dose for immunocompromised individuals, recognizing their heightened vulnerability. Additionally, the FDA granted full approval for the Moderna vaccine in January 2022 for individuals aged 18 and older, following further data confirming its long-term safety and efficacy. This full approval reinforced public confidence and expanded its use beyond emergency conditions.
In conclusion, the Moderna vaccine’s approval and safety data review by the FDA exemplify the balance between urgency and scientific rigor in pandemic response. Its mRNA technology, coupled with robust clinical trial results, positioned it as a reliable and effective vaccine. For individuals, understanding its dosage, side effects, and storage requirements ensures informed decision-making. As vaccination efforts continue, the Moderna vaccine remains a testament to the power of innovation and regulatory oversight in safeguarding public health.
Link Zelle to Huntington Bank: A Step-by-Step Guide
You may want to see also
Explore related products






![]()
Johnson & Johnson vaccine approval and single-dose efficacy
The Johnson & Johnson COVID-19 vaccine stands out as the first single-dose option approved by the FDA for individuals aged 18 and older. This approval, granted in February 2021, marked a significant milestone in the fight against the pandemic, offering a practical alternative to the two-dose regimens of Pfizer and Moderna. The vaccine’s single-dose efficacy was a game-changer, particularly for populations with limited access to healthcare or those hesitant to commit to multiple appointments. Clinical trials demonstrated that the vaccine was 66% effective in preventing moderate to severe COVID-19 globally, rising to 72% in the United States. While these numbers may seem lower than those of mRNA vaccines, they are still highly effective in preventing hospitalization and death, which are the most critical outcomes.
One of the key advantages of the Johnson & Johnson vaccine is its logistical simplicity. Unlike mRNA vaccines, which require ultra-cold storage, this vaccine can be stored at standard refrigerator temperatures for up to three months, making it easier to distribute in remote or resource-limited areas. Additionally, its single-dose administration reduces the burden on healthcare systems and improves compliance, as individuals only need to schedule one appointment. For those with busy schedules or vaccine hesitancy, this convenience factor cannot be overstated. However, it’s important to note that the vaccine uses a viral vector (adenovirus) to deliver genetic material, which has been associated with rare but serious side effects, such as blood clots with low platelets (TTS). These risks are extremely low, occurring in approximately 7 per 1 million vaccinated women aged 18–49, but they underscore the importance of informed decision-making.
Comparatively, the Johnson & Johnson vaccine’s efficacy against variants has been a topic of discussion. While it has shown reduced effectiveness against strains like Delta and Omicron, booster shots have significantly enhanced its protection. In October 2021, the FDA authorized boosters for individuals aged 18 and older who had received the initial dose at least two months prior. Studies found that a booster increased antibody levels four to sixfold, improving protection against severe illness and hospitalization. This highlights the vaccine’s adaptability and the importance of staying updated with evolving public health recommendations. For those who received the Johnson & Johnson vaccine initially, consulting a healthcare provider about booster eligibility is a practical step to ensure continued protection.
From a persuasive standpoint, the Johnson & Johnson vaccine’s single-dose efficacy makes it a compelling choice for specific populations. For example, it is particularly beneficial for individuals with a history of severe allergic reactions to mRNA vaccine components, as it offers a safe alternative. Moreover, its ease of distribution and administration make it ideal for mass vaccination campaigns in underserved communities or during humanitarian crises. While debates about its lower efficacy compared to mRNA vaccines persist, its real-world impact in preventing severe outcomes cannot be overlooked. The vaccine’s approval and subsequent booster recommendations demonstrate the FDA’s commitment to balancing accessibility with safety and efficacy, ensuring that diverse needs are met in the global fight against COVID-19.
Effortlessly Sync Your Bank Account with HoneyBook: A Step-by-Step Guide
You may want to see also
Explore related products






![]()
FDA's role in monitoring post-vaccination side effects and safety
The FDA's approval of a coronavirus vaccine is just the beginning of its role in ensuring public health. Once a vaccine is administered to millions, the agency shifts its focus to post-vaccination surveillance, a critical phase often overlooked by the public. This ongoing monitoring is essential to identify rare side effects that may not have surfaced during clinical trials, which typically involve tens of thousands of participants. For instance, the FDA's Vaccine Adverse Event Reporting System (VAERS) and the Vaccine Safety Datalink (VSD) are key tools in this process, allowing healthcare providers and individuals to report any adverse reactions. These systems are designed to detect signals that might indicate a safety issue, ensuring that the benefits of the vaccine continue to outweigh the risks.
Consider the practical steps involved in this monitoring process. After receiving a COVID-19 vaccine, individuals are advised to stay at the vaccination site for 15–30 minutes to monitor for immediate reactions, such as anaphylaxis, which, although rare, requires prompt medical attention. The FDA recommends that healthcare providers report any severe or unexpected side effects to VAERS, even if they are unsure whether the vaccine caused the event. For the general public, the CDC’s v-safe program offers a user-friendly way to track health status post-vaccination via smartphone, providing real-time data to health authorities. These layered systems ensure that potential safety concerns are identified and addressed swiftly, maintaining public trust in the vaccination program.
A comparative analysis highlights the FDA’s proactive approach in contrast to passive surveillance methods used in some countries. While passive systems rely on voluntary reporting, which can underreport events, the FDA’s active surveillance through VSD leverages electronic health records from large healthcare organizations to systematically analyze data. This method allows for the rapid detection of patterns, such as the rare incidence of thrombosis with thrombocytopenia syndrome (TTS) following the Johnson & Johnson vaccine, which led to updated guidelines restricting its use in certain age groups. Such agility in response underscores the FDA’s commitment to balancing vaccine accessibility with safety.
Persuasively, the FDA’s role in post-vaccination safety is not just regulatory but educational. The agency regularly communicates findings to healthcare professionals and the public, ensuring transparency. For example, after reports of myocarditis in young males following mRNA vaccines, the FDA updated fact sheets and held public meetings to discuss risks and benefits. This open dialogue empowers individuals to make informed decisions, particularly for parents considering vaccination for children aged 5–11, where dosage is adjusted to 10 micrograms per shot compared to 30 micrograms for adults, reflecting the FDA’s tailored approach to safety across age categories.
In conclusion, the FDA’s monitoring of post-vaccination side effects is a dynamic, multi-faceted process that extends far beyond initial approval. Through active surveillance, public engagement, and adaptive guidelines, the agency ensures that vaccines remain a safe and effective tool in combating the pandemic. This ongoing vigilance not only addresses immediate concerns but also builds a foundation for future vaccine development and public health strategies.
Is the AAMC Section Bank Worth Your MCAT Prep Time?
You may want to see also
Frequently asked questions
Yes, the FDA has approved multiple coronavirus vaccines for use in the United States, including Pfizer-BioNTech (Comirnaty) for individuals aged 16 and older.
The FDA granted full approval to the Pfizer-BioNTech COVID-19 vaccine (Comirnaty) on August 23, 2021.
Some COVID-19 vaccines, like Pfizer-BioNTech, are fully FDA-approved, while others, such as Moderna and Johnson & Johnson, remain under Emergency Use Authorization (EUA) for certain age groups or formulations.
Yes, FDA approval confirms that the vaccines meet rigorous standards for safety, efficacy, and manufacturing quality based on extensive clinical trial data and ongoing monitoring.
Yes, the Pfizer-BioNTech vaccine (Comirnaty) is FDA-approved for individuals aged 16 and older, and it is authorized under EUA for children aged 5 and older.